森下研究室の研究活動の紹介
近年の生命科学では、生み出される大規模データを高速に処理し新しい生物学的知見を導くソフトウエアが、重要な役割を担うようになってきています。我々は、コンピュータサイエンスの最先端手法(アルゴリズム・データマイニング・大規模データ処理)を駆使してソフトウエアを研究し、基本的なバイオインフォマティクス・ソフトはできるだけ自分たちの手でつくり広めることを目標にしています。以下のURLを通じて公開しているソフトウエアは世界的に幅広く利用されています。
http://mlab.cb.k.u-tokyo.ac.jp/
1. ゲノム
動機は、いまだ解読されていない生物種のゲノムを解読し、コードされている遺伝子を予測し、他の生物のゲノムと構造を比較し、進化の過程を推測し、生物種に固有の遺伝子や生物種を超えて保存されている遺伝子を同定することにあります。しかし各ステップには新たなソフトウエアの研究開発が待たれています。たとえば、ヒトゲノム配列の解読には多額の研究資金が使われましたが、今後解読が計画されている哺乳類・脊椎動物ゲノムの場合、コストを抑えて正確に解読することが課題です。5億塩基を超えるゲノム配列を高速に解読することは、数千万ピースのジグソーパズルを解くことにたとえられるほど困難な問題です。我々は解読ソフト Ramenを研究開発中ですが、日本が独力で推進したメダカゲノム(約8億塩基)、カイコゲノム(約5億塩基)の解読に活かされ、また国外のゲノムプロジェクトにも利用されつつあります。他には、遺伝子の予測精度を上げるため効率的に転写開始地点を決定する技術を共同研究者と編み出し、進化的に遠くはなれた生物種のゲノムを感度よく比較して低類似度の配列でも見つけ出すアルゴリズムを研究しています。このようにしてゲノムをアノテーションした結果を独自に研究開発したUT Genome Browser を利用して公開しています。
|
|
UT Genome
Browser |

2. トランスクリプトーム
特定の遺伝子に存在する長さ20前後の短い配列で、他の遺伝子やゲノム配列上にハイブリダイズする可能性が殆どない配列を現実的な時間で見つけることができるか? という基本的問題の解決に取組んでいます。この技術はかなり応用の幅が広く、特定の遺伝子の発現を阻害し他の遺伝子は阻害しない siRNA配列の設計、特定の遺伝子だけを絶対定量もしくは相対定量するプライマやオリゴマの設計、転写開始地点の頻度分布を観測する技術、等へ活かされます。我々の技術は改良の余地がありますが世界をリードし、特に siRNA 配列設計サイト siDirect
( http://design.rnai.jp/ ) と 5’SAGE ( http://5sage.gi.k.u-tokyo.ac.jp/ ) は反響を呼んでいます。また企業へも技術移転されています。さらに、得られた発現量データから遺伝子機能を予測するデータマイニングについても研究しています。最近の成果は2001年ACM主催の KDD Cupコンテストでの優勝と、クラス分類とクラスタリングを同時に実行する知識発見手法の提案で、癌関連遺伝子の解析等に応用しています。
3. プロテオーム
数百種のタンパク質から得られる数多くのペプチド断片情報をゲノム上に写像し、もとのタンパク質配列を推測する、いわゆる逆問題の解決に取組んでおり、新しいプロテオーム解析技術の確立を目指しています。
4. フェノーム
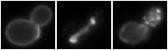
遺伝子の破壊や強制発現が形態にもたらす変化を緻密に定量化すれば、統計学的な根拠から変異体の特徴を記述でき、遺伝子機能の理解に役立つと我々は考えています。そこで出芽酵母のすべての非必須遺伝子の破壊株の顕微鏡写真から、細胞壁・核・アクチンの微小な形態異常をも検出するイメージ処理ソフトウエアを研究開発し、結果を公開しています。またショウジョウバエにおいて遺伝子を強制発現させたとき翅脈にどのような形態変化が起こるかについても同様の分析を行っています。
|
|
上: 出芽酵母 遺伝子破壊株データベースhttp://yeast.gi.k.u-tokyo.ac.jp/ 下: ショウジョウバエ変異体の翅脈解析 |